

Antiandrogens, also known as androgen antagonists or testosterone blockers, are a class of drugs which prevent androgens like testosterone and dihydrotestosterone (DHT) from mediating their biological effects in the body. They act by blocking the androgen receptor (AR) and/or inhibiting or suppressing androgen production. Antiandrogens are one of three types of sex hormone antagonists, the others being antiestrogens and antiprogestogens.
Antiandrogens are used to treat an assortment of androgen-dependent conditions. In males, antiandrogens are used in the treatment of prostate cancer, enlarged prostate, scalp hair loss, overly high sex drive, unusual and problematic sexual urges, and early puberty. In women, antiandrogens are used to treat acne, seborrhea, excessive hair growth, scalp hair loss, and high androgen levels, such as those which occur in polycystic ovary syndrome (PCOS). Antiandrogens are also used as a component of feminizing hormone therapy for transgender women and as puberty blockers in transgender girls.
Side effects of antiandrogens depend on the type of antiandrogen and the specific antiandrogen in question. In any case, common side effects of antiandrogens in men include breast tenderness, breast enlargement, feminization, hot flashes, sexual dysfunction, infertility, and osteoporosis. In women, antiandrogens are much better tolerated, and antiandrogens that work only by directly blocking androgens are associated with minimal side effects. However, because estrogens are made from androgens in the body, antiandrogens that suppress androgen production can cause low estrogen levels and associated symptoms like hot flashes, menstrual irregularities, and osteoporosis in premenopausal women.
There are a few different major types of antiandrogens. These include AR antagonists, androgen synthesis inhibitors, and antigonadotropins. AR antagonists work by directly blocking the effects of androgens, while androgen synthesis inhibitors and antigonadotropins work by lowering androgen levels. AR antagonists can be further divided into steroidal antiandrogens and nonsteroidal antiandrogens; androgen synthesis inhibitors can be further divided mostly into CYP17A1 inhibitors and 5?-reductase inhibitors; and antigonadotropins can be further divided into gonadotropin-releasing hormone analogues (GnRH analogues), progestogens, and estrogens.
Video Antiandrogen
Types and examples
There are several different varieties of antiandrogens, including the following:
- Androgen receptor antagonists: drugs that bind directly to and block the AR. These drugs include the steroidal antiandrogens cyproterone acetate, megestrol acetate, chlormadinone acetate, spironolactone, oxendolone, and osaterone acetate (veterinary) and the nonsteroidal antiandrogens flutamide, bicalutamide, nilutamide, topilutamide, and enzalutamide. Aside from cyproterone acetate and chlormadinone acetate, a few other progestins used in oral contraceptives and/or in menopausal HRT including dienogest, drospirenone, medrogestone, and nomegestrol acetate also have varying degrees of AR antagonistic activity.
- Androgen synthesis inhibitors: drugs that directly inhibit the enzymatic biosynthesis of androgens like testosterone and/or DHT. Examples include the CYP17A1 inhibitors ketoconazole, abiraterone acetate, and seviteronel, the CYP11A1 (P450scc) inhibitor aminoglutethimide, and the 5?-reductase inhibitors finasteride, dutasteride, epristeride, alfatradiol, and saw palmetto extract (Serenoa repens). A number of other antiandrogens, including cyproterone acetate, spironolactone, medrogestone, flutamide, nilutamide, and bifluranol, are also known to weakly inhibit androgen synthesis.
- Antigonadotropins: drugs that suppress the gonadotropin-releasing hormone (GnRH)-induced release of gonadotropins and consequent activation of gonadal androgen production. Examples include GnRH analogues like leuprorelin (a GnRH agonist) and cetrorelix (a GnRH antagonist), progestogens like chlormadinone acetate, cyproterone acetate, gestonorone caproate, medroxyprogesterone acetate, megestrol acetate, osaterone acetate (veterinary), and oxendolone, and estrogens like estradiol, estradiol esters, ethinylestradiol, conjugated equine estrogens (Premarin), diethylstilbestrol, and bifluranol.
Certain antiandrogens combine multiple of the above mechanisms. An example is the steroidal antiandrogen cyproterone acetate, which is a potent AR antagonist, a potent progestogen and hence antigonadotropin, and a weak androgen synthesis inhibitor.
Although the term antiandrogen is generally used to refer specifically to AR antagonists, it may also be used to describe functional antiandrogens like androgen synthesis inhibitors and antigonadotropins, including even estrogens and progestogens. For example, the progestogen and hence antigonadotropin medroxyprogesterone acetate is sometimes described as a steroidal antiandrogen, even though it is not an antagonist of the AR.
Maps Antiandrogen
Medical uses
Antiandrogens are used in the treatment of an assortment of androgen-dependent conditions in both men and women. They are used to treat men with prostate cancer, benign prostatic hyperplasia, pattern hair loss, hypersexuality, and paraphilias, as well as boys with precocious puberty. They have also been used to treat men with priapism. In women, antiandrogens are used to treat acne, seborrhea, hidradenitis suppurativa, hirsutism, and hyperandrogenism. Antiandrogens are also used in transgender women as a component of HRT and as puberty blockers in transgender girls.
Males
Prostate cancer
Androgens like testosterone and particularly DHT are importantly involved in the development and progression of prostate cancer. They act as growth factors in the prostate gland, stimulating cell division and tissue growth. In accordance, therapeutic modalities that reduce androgen signaling in the prostate gland, referred to collectively as androgen deprivation therapy, are able to significantly slow the course of prostate cancer and extend life in men with the disease. Although antiandrogens are effective in slowing the progression of prostate cancer, they are not generally curative, and with time, the disease adapts and androgen deprivation therapy eventually becomes ineffective. When this occurs, other treatment approaches, such as chemotherapy, may be considered.
The most common methods of androgen deprivation therapy currently employed to treat prostate cancer are castration (with a GnRH analogue or orchiectomy), nonsteroidal antiandrogens, and the androgen synthesis inhibitor abiraterone acetate. Castration may be used alone or in combination with one of the other two treatments. When castration is combined with a nonsteroidal antiandrogen like bicalutamide, this strategy is referred to as combined androgen blockade (also known as complete or maximal androgen blockade). Enzalutamide and abiraterone acetate are specifically approved for use in combination with castration to treat advanced castration-resistant prostate cancer. Monotherapy with the nonsteroidal antiandrogen bicalutamide is also used in the treatment of prostate cancer as an alternative to castration with comparable effectiveness but with a different and potentially advantageous side effect profile.
High-dose estrogen was the first functional antiandrogen to be used in the treatment of prostate cancer and was widely employed, but has largely been abandoned for this indication in favor of newer agents with improved safety profiles and fewer feminizing side effects. Cyproterone acetate was developed subsequently to high-dose estrogen and is the only steroidal antiandrogen that has been widely used in the treatment of prostate cancer, but it has largely been replaced by nonsteroidal antiandrogens, which are newer and have greater effectiveness, tolerability, and safety. Bicalutamide, as well as enzalutamide, have largely replaced the earlier nonsteroidal antiandrogens flutamide and nilutamide, which are now little used. The earlier androgen synthesis inhibitors aminoglutethimide and ketoconazole have only limitedly been used in the treatment of prostate cancer due to toxicity concerns and have been replaced by abiraterone acetate.
In addition to active treatment of prostate cancer, antiandrogens are effective as prophylaxis (preventatives) in reducing the risk of ever developing prostate cancer. Antiandrogens have only limitedly been assessed for this purpose, but the 5?-reductase inhibitors finasteride and dutasteride and the steroidal AR antagonist spironolactone have been associated with significantly reduced risk of prostate cancer. In addition, it is notable that prostate cancer is extremely rare in transgender women who have been on HRT for an extended period of time.
Enlarged prostate
The 5?-reductase inhibitors finasteride and dutasteride are used to treat benign prostatic hyperplasia, a condition in which the prostate becomes enlarged and this results in urinary obstruction and discomfort. They are effective because androgens act as growth factors in the prostate gland. The functional antiandrogens oxendolone and gestonorone caproate are also approved in some countries for the treatment of benign prostatic hyperplasia.
Scalp hair loss
5?-Reductase inhibitors like finasteride, dutasteride, and alfatradiol and the topical nonsteroidal AR antagonist topilutamide (fluridil) are approved for the treatment of pattern hair loss, also known as scalp hair loss or baldness. This condition is generally caused by androgens, so antiandrogens can slow or halt its progression.
Hypersexuality and paraphilias
Androgens increase sex drive, and for this reason, antiandrogens are able to reduce sex drive in men. In accordance, antiandrogens are used in the treatment of hypersexuality (excessively high sex drive) and paraphilias (atypical and sometimes societally unacceptable sexual interests) like pedophilia (sexual attraction to children). They have been used to decrease sex drive in sex offenders so as to reduce the likelihood of recidivism (repeat offenses). Antiandrogens used for these indications include cyproterone acetate, medroxyprogesterone acetate, and GnRH analogues.
Long-lasting erections
Antiandrogens are effective in the treatment of recurrent priapism (potentially painful penile erections that last more than four hours).
Females
Skin and hair conditions
Antiandrogens are used in the treatment of androgen-dependent skin and hair conditions including acne, seborrhea, hidradenitis suppurativa, hirsutism, and pattern hair loss in women. All of these conditions are dependent on androgens, and for this reason, antiandrogens are effective in treating them. The most commonly used antiandrogens for these indications are cyproterone acetate and spironolactone. Flutamide has also been studied extensively for such uses, but has fallen out of favor due to its association with hepatotoxicity. Bicalutamide, which has a relatively minimal risk of hepatotoxicity, has been evaluated for the treatment of hirsutism and found to be effective similarly to flutamide and may be used instead of it. In addition to AR antagonists, oral contraceptives containing ethinylestradiol are effective in treating these conditions, and may be combined with AR antagonists.
Antiandrogens generally are not used to treat acne in males due to the high risk of feminization (e.g., gynecomastia) and sexual dysfunction.
High androgen levels
Hyperandrogenism is a condition in women in which androgen levels are excessively and abnormally high. It is commonly seen in women with PCOS, and also occurs in women with intersex conditions like congenital adrenal hyperplasia. Hyperandrogenism is associated with virilization - that is, the development of masculine secondary sexual characteristics like male-pattern facial and body hair growth (or hirsutism), voice deepening, increased muscle mass and strength, and broadening of the shoulders, among others. Androgen-dependent skin and hair conditions like acne and pattern hair loss may also occur in hyperandrogenism, and menstrual disturbances, like amenorrhea, are commonly seen. Although antiandrogens do not treat the underlying cause of hyperandrogenism (e.g., PCOS), they are able to prevent and reverse its manifestation and effects. As with androgen-dependent skin and hair conditions, the most commonly used antiandrogens in the treatment of hyperandrogenism in women are cyproterone acetate and spironolactone. Other antiandrogens, like bicalutamide, may be used alternatively.
Transgender women
Antiandrogens are used to prevent or reverse masculinization and to facilitate feminization in transgender women who are undergoing HRT and who have not undergone sex reassignment surgery or orchiectomy. Besides estrogens, the main antiandrogens that have been used for this purpose are cyproterone acetate, spironolactone, and GnRH analogues. Nonsteroidal antiandrogens like bicalutamide are also used for this indication. In addition to use in transgender women, antiandrogens, mainly GnRH analogues, are used as puberty blockers to prevent puberty in transgender girls until they are older and ready to begin HRT.

Side effects
The side effects of antiandrogens vary depending on the type of antiandrogen - namely whether it is a selective AR antagonist or lowers androgen levels - as well as the presence of off-target activity in the antiandrogen in question. For instance, whereas antigonadotropic antiandrogens like GnRH analogues and cyproterone acetate are associated with pronounced sexual dysfunction and osteoporosis in men, selective AR antagonists like bicalutamide are not associated with osteoporosis and have been associated with only minimal sexual dysfunction. These differences are thought to be related to the fact that antigonadotropins suppress androgen levels and by extension levels of bioactive metabolites of androgens like estrogens and neurosteroids whereas selective AR antagonists similarly neutralize the effects of androgens but leave levels of androgens and hence their metabolites intact (and in fact can even increase them as a result of their progonadotropic effects). As another example, the steroidal antiandrogens cyproterone acetate and spironolactone possess off-target actions including progestogenic, antimineralocorticoid, and/or glucocorticoid activity in addition to their antiandrogen activity, and these off-target activities can result in additional side effects.
In males, the major side effects of antiandrogens are demasculinization and feminization. These side effects include breast pain/tenderness and gynecomastia (breast development/enlargement), reduced body hair growth/density, decreased muscle mass and strength, feminine changes in fat mass and distribution, and reduced penile length and testicular size. In addition, antiandrogens can cause infertility, osteoporosis, hot flashes, sexual dysfunction (including loss of libido and erectile dysfunction), depression, fatigue, anemia, and decreased semen/ejaculate volume in males. Conversely, the side effects of selective AR antagonists in women are minimal. However, antigonadotropic antiandrogens like cyproterone acetate can produce hypoestrogenism, amenorrhea, and osteoporosis in premenopausal women, among other side effects.
A number of antiandrogens have been associated with hepatotoxicity. These include, to varying extents, cyproterone acetate, flutamide, nilutamide, bicalutamide, aminoglutethimide, and ketoconazole. In contrast, spironolactone, enzalutamide, and other antiandrogens are not associated with hepatotoxicity. However, although they do not pose a risk of hepatotoxicity, spironolactone has a risk of hyperkalemia and enzalutamide has a risk of seizures.
In women who are pregnant, antiandrogens can interfere with the androgen-mediated sexual differentiation of the genitalia and brain of male fetuses. This manifests primarily as ambiguous genitalia - that is, undervirilized or feminized genitalia which, anatomically, are a cross between a penis and a vagina - and theoretically also as femininity and homosexuality. As such, antiandrogens are teratogens, and women who are pregnant should not be treated with an antiandrogen. Moreover, women who can or may become pregnant are strongly recommended to take an antiandrogen only in combination with proper contraception.

Mechanism of action
Androgen receptor antagonists
AR antagonists act by directly binding to and competitively displacing androgens like testosterone and DHT from the AR, thereby preventing them from activating the receptor and mediating their biological effects. AR antagonists are classified into two types, based on chemical structure: steroidal and nonsteroidal. Steroidal AR antagonists are structurally related to steroid hormones like testosterone and progesterone, whereas nonsteroidal AR antagonists are not steroids and are structurally distinct. Steroidal AR antagonists tend to have off-target hormonal actions due to their structural similarity to other steroid hormones. In contrast, nonsteroidal AR antagonists are selective for the AR and have no off-target hormonal activity. For this reason, they are sometimes described as "pure" antiandrogens.
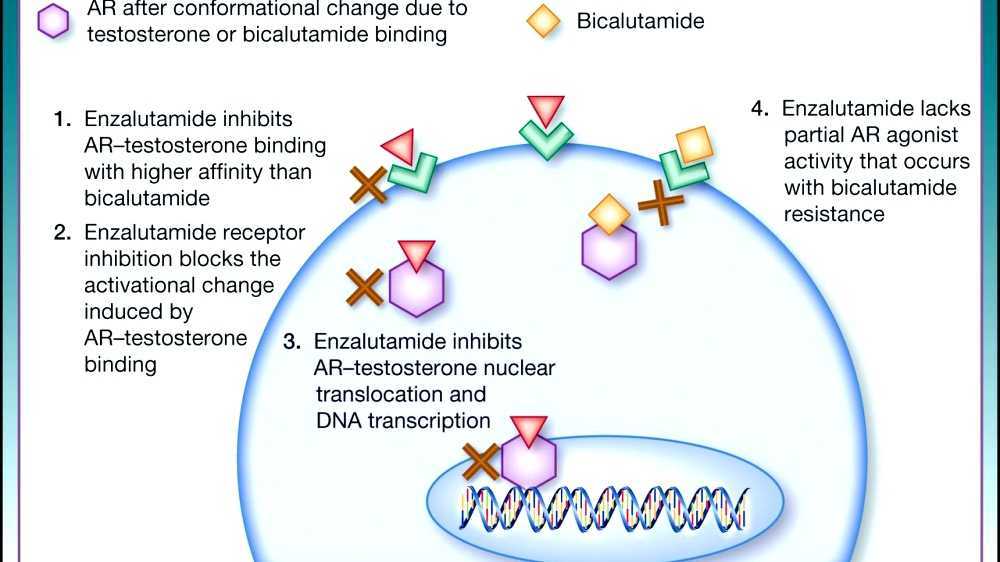
Although they are described as antiandrogens and indeed show only such effects generally, most or all steroidal AR antagonists are actually not silent antagonists of the AR but rather are weak partial agonists and are able to activate the receptor in the absence of more potent AR agonists like testosterone and DHT. This may have clinical implications in the specific context of prostate cancer treatment. As an example, steroidal AR antagonists are able to increase prostate weight and accelerate prostate cancer cell growth in the absence of more potent AR agonists, and spironolactone has been found to accelerate progression of prostate cancer in case reports. In addition, whereas cyproterone acetate produces ambiguous genitalia via feminization in male fetuses when administered to pregnant animals, it has been found to produce masculinization of the genitalia of female fetuses of pregnant animals. In contrast to steroidal AR antagonists, nonsteroidal AR antagonists are silent antagonists of the AR and do not activate the receptor. This may be why they have greater efficacy than steroidal AR antagonists in the treatment of prostate cancer and is an important reason as to why they have largely replaced them for this indication in medicine.
AR antagonists may not bind to or block membrane androgen receptors (mARs), which are distinct from the classical nuclear AR. However, the mARs do not appear to be involved in masculinization. This is evidenced by the perfectly female phenotype of women with complete androgen insensitivity syndrome. These women have a 46,XY karyotype (i.e., are genetically "male") and high levels of androgens but possess a defective AR and for this reason never masculinize. They are described as highly feminine, both physically as well as mentally and behaviorally.
N-Terminal domain antagonists
N-Terminal domain AR antagonists are a new type of AR antagonist which, unlike all currently marketed AR antagonists, bind to the N-terminal domain (NTD) of the AR rather than the ligand-binding domain (LBD). Whereas conventional AR antagonists bind to the LBD of the AR and competitively displace androgens, thereby preventing them from activating the receptor, AR NTD antagonists bind covalently to the NTD of the AR and prevent protein-protein interactions subsequent to activation that are required for transcriptional activity. As such, they are non-competitive and irreversible antagonists of the AR. Examples of AR NTD antagonists include bisphenol A diglycidyl ether (BADGE) and its derivatives EPI-001, ralaniten (EPI-002), and ralaniten acetate (EPI-506). AR NTD antagonists are under investigation for the potential treatment of prostate cancer, and it is thought that they may have greater efficacy as antiandrogens relative to conventional AR antagonists. In accordance with this notion, AR NTD antagonists are active against splice variants of the AR, which conventional AR antagonists are not, and AR NTD antagonists are immune to gain-of-function mutations in the AR LBD that convert AR antagonists into AR agonists and which commonly occur in prostate cancer.
Androgen receptor degraders
Selective androgen receptor degraders (SARDs) are another new type of antiandrogen that has recently been developed. They work by enhancing the degradation of the AR, and are analogous to selective estrogen receptor degraders (SERDs) like fulvestrant (a drug used to treat estrogen receptor-positive breast cancer). Similarly to AR NTD antagonists, it is thought that SARDs may have greater efficacy than conventional AR antagonists, and for this reason, they are under investigation for the treatment of prostate cancer. An example of a SARD is dimethylcurcumin (ASC-J9), which is under development as a topical medication for the potential treatment of acne. SARDs like dimethylcurcumin differ from conventional AR antagonists and AR NTD antagonists in that they may not necessarily bind directly to the AR.
Androgen synthesis inhibitors
Androgen synthesis inhibitors are enzyme inhibitors which prevent the biosynthesis of androgens. This process occurs mainly in the gonads and adrenal glands. These drugs include aminoglutethimide, ketoconazole, and abiraterone acetate. Aminoglutethimide inhibits cholesterol side-chain cleavage enzyme, also known as P450scc or CYP11A1, which is responsible for the conversion of cholesterol into pregnenolone and by extension the production of all steroid hormones, including the androgens. Ketoconazole and abiraterone acetate are inhibitors of the enzyme CYP17A1, also known as 17?-hydroxylase/17,20-lyase, which is responsible for the conversion of pregnane steroids into androgens, as well as the conversion of mineralocorticoids into glucocorticoids. Because these drugs all prevent the formation of glucocorticoids in addition to androgens, they must be combined with a glucocorticoid like prednisone to avoid adrenal insufficiency. A newer drug which is currently under development for the treatment of prostate cancer, seviteronel, is selective for inhibition of the 17,20-lyase functionality of CYP17A1, and for this reason, unlike earlier drugs, does not require concomitant treatment with a glucocorticoid.
5?-Reductase inhibitors
5?-Reductase inhibitors such as finasteride and dutasteride are inhibitors of 5?-reductase, an enzyme that is responsible for the formation of DHT from testosterone. DHT is between 2.5- and 10-fold more potent than testosterone as an androgen and is produced in a tissue-selective manner based on expression of 5?-reductase. Tissues in which DHT is formed at a high rate include the prostate gland, skin, and hair follicles. In accordance, DHT is involved in the pathophysiology of benign prostatic hyperplasia, pattern hair loss, and hirsutism, and 5?-reductase inhibitors are used to treat these conditions. Because 5?-reductase inhibitors selectively prevent the formation of DHT and do not affect testosterone levels, and because DHT is important as an endgenous androgen only in select tissues, 5?-reductase inhibitors have minimal side effects in both men and women, unlike other antiandrogens.
Antigonadotropins
Antigonadotropins are drugs which suppress the GnRH-mediated secretion of gonadotropins from the pituitary gland. Gonadotropins include luteinizing hormone (LH) and follicle-stimulating hormone (FSH) and are peptide hormones that signal the gonads to produce sex hormones. By suppressing gonadotropin secretion, antigonadotropins suppress gonadal sex hormone production and by extension circulating androgen levels. GnRH analogues, including both GnRH agonists and GnRH antagonists, are powerful antigonadotropins that are able to suppress androgen levels by 95% in men. In addition, estrogens and progestogens are antigonadotropins via exertion of negative feedback on the hypothalamic-pituitary-gonadal axis. High-dose estrogens are able to suppress androgen levels to castrate levels in men similarly to GnRH analogues, while high-dose progestogens are able to suppress androgen levels by up to approximately 70 to 80% in men.
Examples of GnRH agonists include leuprorelin (leuprolide) and goserelin, while an example of a GnRH antagonist is cetrorelix. Estrogens that are or that have been used as antigonadotropins include estradiol, estradiol esters like estradiol valerate, estradiol undecylate, and polyestradiol phosphate, conjugated equine estrogens, ethinylestradiol, diethylstilbestrol (no longer widely used), and bifluranol. Progestogens that are used as antigonadotropins include chlormadinone acetate, cyproterone acetate, gestonorone caproate, medroxyprogesterone acetate, megestrol acetate, and oxendolone.
Miscellaneous
Blood proteins
In addition to their antigonadotropic effects, estrogens are also functional antiandrogens by decreasing free concentrations of androgens via increasing the hepatic production of sex hormone-binding globulin (SHBG) and by extension circulating SHBG levels. Combined oral contraceptives containing ethinylestradiol have been found to increase circulating SHBG levels by 2- to 4-fold in women and to reduce free testosterone concentrations by 40 to 80%. However, combined oral contraceptives containing the particularly androgenic progestin levonorgestrel have been found to increase SHBG levels by only 50 to 100%, which is likely due to the fact that activation of the AR in the liver has the opposite effect of estrogen and suppresses production of SHBG. Levonorgestrel and certain other 19-nortestosterone progestins used in combined oral contraceptives like norethisterone also directly bind to and displace androgens from SHBG, which may additionally antagonize the functional antiandrogenic effects of ethinylestradiol. In men, a study found that treatment with a relatively low dosage of 20 ?g/day ethinylestradiol for 5 weeks increased circulating SHBG levels by 150% and, due to the accompanying decrease free testosterone levels, increased total circulating levels of testosterone by 50% (via reduced negative feedback by androgens on the hypothalamic-pituitary-gonadal axis).
Immunization
Ovandrotone albumin (Fecundin, Ovastim) and Androvax (androstenedione albumin) are immunogens and vaccines against androstenedione that are used in veterinary medicine to improve fecundity (reproductive rate) in ewes (adult female sheep). The generation of antibodies against androstenedione by these agents is thought to decrease circulating levels of androstenedione and its metabolites (e.g., testosterone and estrogens), which in turn increases the activity of the hypothalamic-pituitary-gonadal axis via reduced negative feedback and increases the rate of ovulation, resulting in greater fertility and fecundity.

History
Antigonadotropins like estrogens and progestogens were both first introduced in the 1930s. The beneficial effects of androgen deprivation via surgical castration or high-dose estrogen therapy on prostate cancer were discovered in 1941. AR antagonists were first discovered in the early 1960s. The steroidal antiandrogen cyproterone acetate was discovered in 1961 and introduced in 1973 and is often described as the first antiandrogen to have been marketed. However, spironolactone was introduced in 1959, although its antiandrogen effects were not recognized or taken advantage of until later and were originally an unintended off-target action of the drug. In addition to spironolactone, chlormadinone acetate and megestrol acetate are steroidal antiandrogens that are weaker than cyproterone acetate but were also introduced earlier, in the 1960s. Other early steroidal antiandrogens that were developed around this time but were never marketed include benorterone (SKF-7690; 17?-methyl-B-nortestosterone), BOMT (Ro 7-2340), cyproterone (SH-80881), and trimethyltrienolone (R-2956).
The nonsteroidal antiandrogen flutamide was first reported in 1967. It was introduced in 1983 and was the first nonsteroidal antiandrogen to be marketed. Another early nonsteroidal antiandrogen, DIMP (Ro 7-8117), which is structurally related to thalidomide and is a relatively weak antiandrogen, was first described in 1973 and was never marketed. Flutamide was followed by nilutamide in 1989 and bicalutamide in 1995. In addition to these three drugs, which have been regarded as first-generation nonsteroidal antiandrogens, the second-generation nonsteroidal antiandrogen enzalutamide was introduced in 2012. It differs from the earlier nonsteroidal antiandrogens namely in that it is much more efficacious in comparison.
The androgen synthesis inhibitors aminoglutethimide and ketoconazole were first marketed in 1960 and 1977, respectively, and the newer drug abiraterone acetate was introduced in 2011. GnRH analogues were first introduced in the 1980s. The 5?-reductase inhibitors finasteride and dutasteride were introduced in 1992 and 2002, respectively.

Research
Topical administration
There has been much interest and effort in the development of topical AR antagonists to treat androgen-dependent conditions like acne and pattern hair loss in males. Unfortunately, whereas systemic administration of antiandrogens is very effective in treating these conditions, topical administration has disappointingly been found generally to possess limited and only modest effectiveness, even when high-affinity steroidal AR antagonists like cyproterone acetate and spironolactone have been employed. Moreover, in the specific case of acne treatment, topical AR antagonists have been found to be greatly inferior in effectiveness compared to established treatments like benzoyl peroxide and antibiotics.
A variety of AR antagonists have been developed for topical use but have not completed development and hence have never been marketed. These include the steroidal AR antagonists cortexolone 17?-propionate, cyproterone, rosterolone, and topterone and the nonsteroidal AR antagonists cioteronel, inocoterone acetate, RU-22930, RU-58642, and RU-58841. However, one topical AR antagonist, topilutamide (fluridil), has been introduced in a few European countries for the treatment of pattern hair loss in men. In addition, a topical 5?-reductase inhibitor and weak estrogen, alfatradiol, has also been introduced in some European countries for the same indication, although its effectiveness is controversial. Spironolactone has been marketed in Italy in the form of a topical cream under the brand name Spiroderm for the treatment of acne and hirsutism, but this formulation was discontinued and hence is no longer available.

See also
- Anabolic-androgenic steroid
- Androgen insensitivity syndrome
- Antiandrogens in the environment
- Selective androgen receptor modulator
- List of antiandrogens
- List of steroidal antiandrogens

References
Source of article : Wikipedia
